Published at 2020-08-18 22:05
Author:zhixy
View:2928
The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215:403-410. DOI: 10.1016/S0022-2836(05)80360-2 Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., & Madden T.L. (2008) BLAST: architecture and applications. BMC Bioinformatics 10:421. DOI: 10.1186/1471-2105-10-421
DIAMOND is a sequence aligner for protein and translated DNA searches, designed for high performance analysis of big sequence data. The key features are:
Buchfink B, Xie C, Huson DH, Fast and sensitive protein alignment using DIAMOND, Nature Methods 12, 59-60 (2015). DOI: 10.1038/nmeth.3176
MMseqs2 (Many-against-Many sequence searching) is a software suite to search and cluster huge protein and nucleotide sequence sets. MMseqs2 can run 10000 times faster than BLAST. At 100 times its speed it achieves almost the same sensitivity. It can perform profile searches with the same sensitivity as PSI-BLAST at over 400 times its speed.
Steinegger M and Soeding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology, 2017 DOI: 10.1038/nbt.3988.
Steinegger M and Soeding J. Clustering huge protein sequence sets in linear time. Nature Communications, 2018 DOI: 10.1038/s41467-018-04964-5.
Mirdita M, Steinegger M and Soeding J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics, 2019 DOI: 10.1093/bioinformatics/bty1057.
By using two genomes of Staphylococcus aureus (GCA_003010475.1 and GCA_003031485.1) as test data, the performances of these three programs were compared under the same hardware conditions, and results are as follows.
(base) [user@server ~]# time blastp -query query.fas -db database -outfmt 6 -out blast.out -evalue 1e-5
real 1m5.989s
user 1m5.675s
sys 0m0.208s
(base) [user@server ~]# time diamond blastp --more-sensitive --evalue 1e-5 -p 1 -q query.fas -d database.dmnd -f 6 --quiet -o diamond.out
real 0m18.520s
user 0m18.470s
sys 0m0.047s
(base) [user@server ~]# time mmseqs easy-search -s 5.7 -e 1e-5 --threads 1 -v 1 query.fas GCA_003010475.1.fasta mmseqs.out tmp
real 0m23.999s
user 0m23.551s
sys 0m0.448s
All hits from Blast (9889 hits), Diamond (6632 hits) and MMseqs (9039 hits) outputs were extracted and subjected to make following venn graph. Blast and MMseqs shared more hits, and Diamond found less hits.
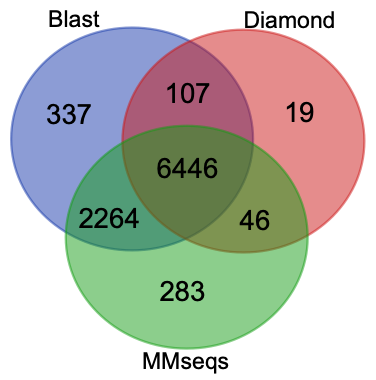
If all hits with identity < 50% were removed, the situation changed.
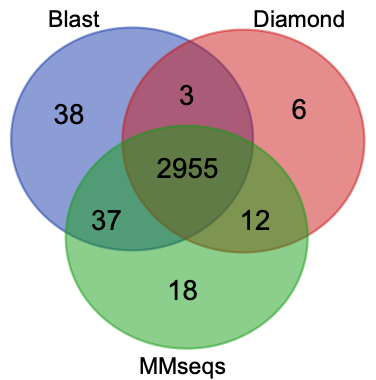
The percentage of overlapping among three outputs increased dramatically (from 67.83% to 96.28%). It indicated that Blast is most sensitive, MMseqs is similar to Blast. However, to the hits with higher identity, three softwares have similar performance.

