Published at 2020-06-12 16:52
Author:zhixy
View:1196
对于一个系统发育群体,如一个属级分类单元,自其共同祖先开始,经过长期的物种分化,形成了当前的物种多样性。系统发育分析的目的,即是借助分子进化的模型来估计(重建)自共同祖先以来的演化历史,并用二叉树的形式表达出来。在有根二叉树上,离所有末端分枝(代表各物种)最远的根分枝,即为共同祖先;而树上的所有中间节点,代表物种分化的中间状态(部分物种的共同祖先)。
物种分化的过程中,细胞内含的基因组,也会因基因获得、基因丢失和新基因的产生等进化事件的影响,而使基因组成(gene content)发生变化。基因组成的变化形式上,将表现为基因家族的演替(gene family turnover)。假设从祖先A到祖先B的演化仅有获得和丢失两种事件,那么获得速率、丢失速率共同决定了在祖先A的基因组成的基础上,演变成祖先B的基因组成的情形。
基因家族的演替分析,就是在一个已知的系统发育历史(物种树)前提下,来估计基因组成进化事件的发生速率,并借此估计各祖先节点的基因组成,依此来勾画在物种分化过程中基因组或关心的部分基因家族的变化规律。

BadiRate是一个用perl语言开发的软件包,用于在系统发育背景下,通过基于似然的方法(或简约法)来估计):
下载源码包,解压后,即可运行主程序BadiRate.pl。
运行环境中perl库,需要有满足运行BadiRate.pl依赖的软件包。
BadiRate需要一颗能够反映物种/基因组关系的进化树,通常选用SuperMatrix树。
物种树中的末端分枝标签(物种名/基因组名标记)必须与下述基因家族数据表中(自第二列以后)的表头标签整体对应(忽略顺序)。
在利用OrthoFinder等软件进行基因家族分析后,得到的主要结果为,在所有基因组中存在的基因组家族,以及这些基因家族中在各基因组中的分布(基因拷贝)。结果可以转换为下图所示的形式。
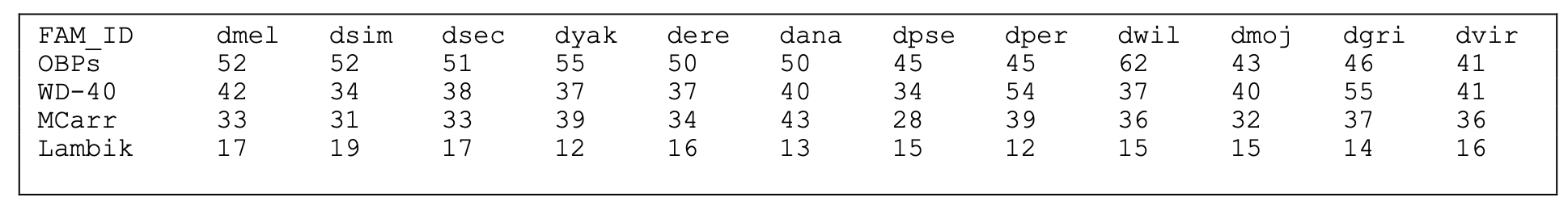
其中,第一列为基因家族的名称或ID,自第二列以后,每列呈现一个基因组的数据,即相应基因家族在该基因组中存在的基因拷贝数。
以上表格可存为
tab键分隔的纯文本文件;需要注意的是:第一列表头必须为FAM_ID
利用BadiRate进行基因家族演替分析,详细地过程可参考其用户文档。
在开始操作之前,需要了解以下细节。
BadiRate的基因家族演替模型(通过-rmodel参数设置)包括:
BadiRate可以模拟基因家族的演替,并假设不同的系统发育分枝上有不同的演替速率参数(通过-bmodel设置)。有以下三种情形:
BadiRate估计速率的统计方法包括(通过-ep设置,通常默认即可):
BadiRate有两项关键输入:
-treefile引入-sizefile引入通过-bmodel可为特定分枝设定不同的演替速率参数,那么在用户给定的物种树上,如何标记那些分枝呢?
首先需要知道BadiRate在分析计算时,会给用户引入的物种树每个分枝做编号标记。在BadiRate示例中,运行:
user@server:~ # perl BadiRate.pl –treefile examples/droso.12sp.tamura.nwk –sizefile examples/obp_all.12sp.tsv –print_ids
((((((dmel_1:5,(dsec_2:1,dsim_3:1)4:4)5:8,(dyak_6:10,dere_7:10)8:3)9:31,dana_10:44)11:11,(dpse_12:1,dper_13:1)14:54)15:7,dwil_16:62)17:1,((dmoj_18:31,dvir_19:31)20:12,dgri_21:43)22:20)23;
而输入的droso.12sp.tamura.nwk为:
((((((dmel:5,(dsec:1,dsim:1):4):8,(dyak:10,dere:10):3):31,dana:44):11,(dpse:1,dper:1):54):7,dwil:62):1,((dmoj:31,dvir:31):12,dgri:43):20);
不难看出每个分枝,都用数字索引做了标记,区别是:内部分枝直接用数字,而末端分枝在数字前加了下划线。可视化效果如下:
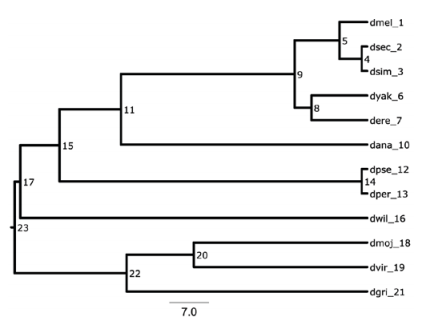
树上的分枝,可用左节点->右节点来表示,例如4->2。在给-bmodel设定取值时,用:号连接分枝表示,这些分枝有相同的速率参数;用_号连接分枝表示,这些分枝有不相同的速率参数。
例如:"17->16_4->2:4->3",表示分枝4->2和4->3为一组(有相同速率参数),而分枝17->16为另一组,与4->2和4->3的速率参数不同,最后所有未明确定义的分枝都归未第三组。因此这样的分枝特异模型的分析命令如下:
user@server:~ # perl BadiRate.pl –treefile examples/droso.12sp.tamura.nwk –sizefile examples/obp_all.12sp.tsv –bmodel "17->16_4->2:4->3" #注意双引号
user@server:~ # perl BadiRate.pl –treefile examples/droso.12sp.tamura.nwk –sizefile examples/obp_all.12sp.tsv –bmodel "17->16_4->2:4->3" -rmodel BDI -anc
-anc参数,为内部节点报告基因家族数据,以及每个谱系中的最小基因获得和丢失数目
上例中,我们用BDI模型模拟基因家族的演替,如前所述BadiRate支持5种模型,再加不同的分枝模型,因此可以演变出多种分析方式。最优模型的选择,涉及模型检验问题。这里我们可以用数据结果中提供的似然值,进行赤池信息量(AIC = 2K – 2ln(L),K为参数数量)的比较来选择最优模型。
关于参数的数量K,我们可以这样计算。以下是基于BGI-BR-ML的一个实例结果:
211 ##Family Turnover Rates
212 #Likelihood: -694741.01170948
213 #Branch_Group Birth Death Innovation
214 0 0.0003560 0.0071065 0.0026219
215 1 0.0000303 0.0030381 0.0008234
216 2 0.0001156 0.0030175 0.0009618
217 3 0.0000012 0.0017299 0.0002489
该实例中,分枝模型-bmodel被分为4种不同的分枝组,演替模型-rmodel设定为BGI,因此有3个速率需要估计,因此共有12个参数需要估计,所以K=12。
AIC最小的为最优模型
Librado P, Vieira FG, and Rozas J., 2012. BadiRate: Estimating Family Turnover Rates by Likelihood-Based Methods. Bioinformatics 28:279-281. DOI: 10.1093/bioinformatics/btr623

